Every month protein spotlight from Expasy highlights a protein.The archive can be found here.
This month is the month for 2 proteins that has ability to make blood group A, B, AB to universal donor blood groups. There are in fact 2 molecules one for blood group A and one for blood group B. These are 2 bacterial glycosidases – α-N-acetylgalactosaminidase and α-1,3-galactosidase A.
As the blood groups are identified by the presence of an antigen on its surface that makes it either A, B or AB. The O group does not have any known antigen. This particular antigen is determined by a carbohydrate structure on the end of oligosaccharide chains on glycoproteins and glycolipids lodged in the cell’s membrane. Blood group A is defined by a terminal α-1,3-linked N-acetylgalactosamine (GalNAc). Blood group B is defined by a terminal α-1,3-linked galactose (Gal). And blood group O (which should really be pronounced ‘zero’) lacks both of these monosaccharides but instead is defined by an α-1,2-linked fucose. If there was a way of wiping off the monosaccharide entities (GalNAc and Gal) on A cells and B cells respectively, then blood groups A and B could be converted into the universal blood group O. What scientists then needed to find was something very small which could snap off the monosaccharide tips: an enzyme of some sort.
α-N-acetylgalactosaminidase and α-1,3-galactosidase A – which have the power to cleave and discard the GalNAc and Gal entities thus converting a red blood cell into a cell with neither a recognisable A nor B blood group antigen. In other words, they can convert A and B blood groups into a universal O blood group. Alpha-N-acetylgalactosaminidase is known in greater molecular detail than its homolog α-1,3-galactosidase A. Surprisingly, unlike other glycosidases of the same family, it needs the help of a cofactor (NAD+), which is nestled in the depths of a narrow tunnel, to cleave the GalNAc monosaccharide. Another part of the enzyme forms a crater which is large enough to accommodate an antigen from which the monosaccharide tip can be cleaved.
Thursday, October 30, 2008
Tuesday, October 28, 2008
When under attack, plants can signal microbial friends for help
A new research finding at University Delaware proves that plants are not just sitting ducks, they react in many different ways to an external attack. The full story can be found here
Wednesday, October 22, 2008
Substantial biases in ultra-short read data sets from high-throughput DNA sequencing
This paper appeared in September issue of Nucleic Acid Research(September; 36(16): e105.)
They generated and analyzed two Illumina 1G ultra-short read data sets, i.e. 2.8 million 27mer reads from a Beta vulgaris genomic clone and 12.3 million 36mers from the Helicobacter acinonychis genome. They found that error rates range from 0.3% at the beginning of reads to 3.8% at the end of reads. Wrong base calls are frequently preceded by base G. Base substitution error frequencies vary by 10- to 11-fold, with A > C transversion being among the most frequent and C > G transversions among the least frequent substitution errors.
Sequencing Stratergies
To achieve high throughput, the new approaches apply different strategies. 454 Life Sciences has adapted pyrosequencing to a microbead format to sequence 400 000 DNA fragments simultaneously, resulting in a per-run dataset of 100 Mbp with reads averaging 250 bp. SOLiD(Applied Biosystems’ Sequencing by Oligonucleotide Ligation and Detection) sequencing also uses templates immobilized onto microbeads. Here, the sequence of the template DNA is decoded by ligation assays involving oligonucleotides labeled with different fluorophores. The SOLiD read length is currently 25–35 bases, and 2–3 Gbp of data can be collected during an 8-day run. Solexa sequencing is based on amplifying single molecules attached to the surface of a flow cell to generate clusters of identical molecules, followed by sequencing using fluorophore-labeled reversible chain terminators. Solexa sequencing proceeds a base at a time and read length depends on the number of sequencing cycles. Current Illumina sequencing instrumentation achieves read lengths of 36 bases. The Solexa flow cell is composed of eight separately loadable lanes. Since each lane has a capacity of about 5 million reads, > 40 million reads can be generated in a run of 3 days, equivalent to > 1.3 Gbp.
This is quite a finding, since many of us are rapidly moving into high throughput sequencing...
They generated and analyzed two Illumina 1G ultra-short read data sets, i.e. 2.8 million 27mer reads from a Beta vulgaris genomic clone and 12.3 million 36mers from the Helicobacter acinonychis genome. They found that error rates range from 0.3% at the beginning of reads to 3.8% at the end of reads. Wrong base calls are frequently preceded by base G. Base substitution error frequencies vary by 10- to 11-fold, with A > C transversion being among the most frequent and C > G transversions among the least frequent substitution errors.
Sequencing Stratergies
To achieve high throughput, the new approaches apply different strategies. 454 Life Sciences has adapted pyrosequencing to a microbead format to sequence 400 000 DNA fragments simultaneously, resulting in a per-run dataset of 100 Mbp with reads averaging 250 bp. SOLiD(Applied Biosystems’ Sequencing by Oligonucleotide Ligation and Detection) sequencing also uses templates immobilized onto microbeads. Here, the sequence of the template DNA is decoded by ligation assays involving oligonucleotides labeled with different fluorophores. The SOLiD read length is currently 25–35 bases, and 2–3 Gbp of data can be collected during an 8-day run. Solexa sequencing is based on amplifying single molecules attached to the surface of a flow cell to generate clusters of identical molecules, followed by sequencing using fluorophore-labeled reversible chain terminators. Solexa sequencing proceeds a base at a time and read length depends on the number of sequencing cycles. Current Illumina sequencing instrumentation achieves read lengths of 36 bases. The Solexa flow cell is composed of eight separately loadable lanes. Since each lane has a capacity of about 5 million reads, > 40 million reads can be generated in a run of 3 days, equivalent to > 1.3 Gbp.
This is quite a finding, since many of us are rapidly moving into high throughput sequencing...
Wednesday, October 15, 2008
Working with a 64 bit machine


I thought I will write this in my note book, but instead I ended up putting it on my blog site! This problem has taken a lot of my working time to solve. Recently I have been moving our entire web portal from a 32 bit machine to a 64 bit machine. The data transfer was quite smooth except for some small troubles here and there - like with PHP versions. In older PHP versions, I did not need to write 'php' near the opening tag(
I have written a C program to calculate the Fickett score as well as the coding potential of a given sequence. Then plot the values on fly using perl GD graphics module. To my surprise, the same GD code plotted like Fig-1 in 32 bit machine where as in 64 bit machine the output was a straight line(Fig-2). I tried to compare the computed text output in 2 machines. Although they were not identical but they had quite good similarity. What the heck. Even I got the numerical output and plotted on 32 bit machine still got figures like the first one. Then on a quick look I found that the newer bioperl module(perl -MGD -e 'print $GD::VERSION'; 2.41) is unable to plot anything when there is a number like '-inf'. This is handled very well with older(2.35) GD packages. Apart from this I was also wondering how 'atof' function works in C. Earlier, the codon usage table was written where there was no '0' preceeding the decimal point (like 0.02344). The array that reads this value is just an array of size 5 and I used fscanf function to read this.My syntax was:
(fscanf(fp,"%s",store[i])!=EOF) . The file was something like this :
GCA .0200 GCC .019 GCG .0238 GCT .0255
TGC .0104 TGT .008 GAC .0356 GAT .022
GAA .0234 GAG .0427 TTC .017 TTT .0213
GGA .014 GGC .022 GGG .009 GGT .0178
No matter whatever I did, fscanf always mis-behaved when it is reading a float as a string (with syntax %s).So, instead of reading .0200 and GCC as 2 different variables it sometimes read .0200GCC as one variable and GCC as another. It practically did not ignore the space in between. So, when I converted the string .0200GCC to a float using atof, it produced a different result than just .0200. As a result of which I would get values like -inf when the log likelihood values are calculated, where it should have been some negative integer. Now having learn t this lesson, I would suggest anyone reading a mixed file of characters and float or any other data types to read them any other way then to read it as string. For this particular case something like
(fscanf(fp,"%s%f",tmp,&val) !=EOF) would be more than appropriate..
Monday, October 06, 2008
Retrotransposons Revisited: The Restraint and Rehabilitation of Parasites
Retrotransposons Revisited: The Restraint and Rehabilitation of Parasites - A review paper appeared in this weeks cell. This paper describes most intricate details about the essence of retrotransposons.
Authors:
John L. Goodier1, Corresponding Author Contact Information, E-mail The Corresponding Author and Haig H. Kazazian Jr.1
1Department of Genetics, University of Pennsylvania School of Medicine, 415 Curie Boulevard, Philadelphia, PA 19104, USA
Available online 2 October 2008
Here is some excerpts of the paper:
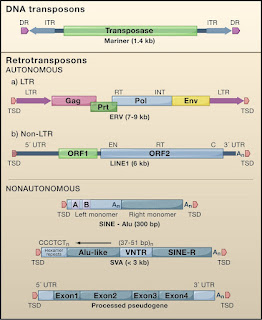
Retroposons mainly the SINES(Short Interspersed Nuclear Elements), LINES(Long Interspersed Nuclear Elements) are endogenous retroviruses, make up roughly 40% of the mammalian genome and have played an important role in genome evolution. In human genome only a small percentage of these LINES(L1s), and SINES are capable of moving into new locations and occasionally causing disease.
There are 2 major groups of such jumping genes. Class II elements or DNA transpososns comprise about 3% of the human genome and most move by a copy paste mechanism. Class I elements comprise of three groups, all moving in a copy paste manner involving reverse transcription of an RNA intermediate and insertion of its cDNA copy or a new site in the genome. Retroviral like or long terminal Reeat(LTR) retrotranspososns include endogenous retroviruses, relics of past rounds of germline infection by viruses that lost their ability to reinfect and become trapped in the genome. These elelments undergo reverse transcription in virus like particles by a complex multistep process. The transposition process for non-LTR retrotransposons is fundamentally different RNA copies of these ellments are likely carried back into the nucleus where their reverse transcription and integration occur in a single step on the DNA itself.
Authors:
John L. Goodier1, Corresponding Author Contact Information, E-mail The Corresponding Author and Haig H. Kazazian Jr.1
1Department of Genetics, University of Pennsylvania School of Medicine, 415 Curie Boulevard, Philadelphia, PA 19104, USA
Available online 2 October 2008
Here is some excerpts of the paper:
Retroposons mainly the SINES(Short Interspersed Nuclear Elements), LINES(Long Interspersed Nuclear Elements) are endogenous retroviruses, make up roughly 40% of the mammalian genome and have played an important role in genome evolution. In human genome only a small percentage of these LINES(L1s), and SINES are capable of moving into new locations and occasionally causing disease.
There are 2 major groups of such jumping genes. Class II elements or DNA transpososns comprise about 3% of the human genome and most move by a copy paste mechanism. Class I elements comprise of three groups, all moving in a copy paste manner involving reverse transcription of an RNA intermediate and insertion of its cDNA copy or a new site in the genome. Retroviral like or long terminal Reeat(LTR) retrotranspososns include endogenous retroviruses, relics of past rounds of germline infection by viruses that lost their ability to reinfect and become trapped in the genome. These elelments undergo reverse transcription in virus like particles by a complex multistep process. The transposition process for non-LTR retrotransposons is fundamentally different RNA copies of these ellments are likely carried back into the nucleus where their reverse transcription and integration occur in a single step on the DNA itself.
Friday, October 03, 2008
Classic Watson Creek one page paper

Molecular Structure Of Nucleic Acids
A Structure for Deoxyribose Nucleic Acid
J.D. Watson, and F.H.C. Crick
Medical Research Council Unit for the Study of the Molecular Structure of Biological Systems, Cavendish Laboratory, Cambridge. April 2.
We wish to suggest a structure for the salt of deoxyribose nucleic acid (D.N.A.). This structure has novel features which are of considerable biological interest.
A structure for nucleic acid has already been proposed by Pauling and Corey (1). They kindly made their manuscript available to us in advance of publication. Their model consists of three intertwined chains, with the phosphates near the fibre axis, and the bases on the outside. In our opinion, this structure is unsatisfactory for two reasons: 1) We believe that the material which gives the X-ray diagrams is the salt, not the free acid. Without the acidic hydrogen atoms it is not clear what forces would hold the structure together, especially as the negatively charged phosphates near the axis will repel each other. 2) Some of the van der Waals distances appear to be too small.
Another three-chain structure has also been suggested by Fraser (in the press). In his model the phosphates are on the outside and the bases on the inside, linked together by hydrogen bonds. This structure as described is rather ill-defined, and for this reason we shall not comment on it.
We wish to put forward a radically different structure for the salt of deoxyribose nucleic acid. This structure has two helical chains each coiled round the same axis (see diagram). We have made the usual chemical assumptions, namely, that each chain consists of phosphate diester groups joining ß-D-deoxyribofuranose residues with 3',5' linkages. The two chains (but not their bases) are related by a dyad perpendicular to the fibre axis. Both chains follow right-handed helices, but owing to the dyad the sequences of the atoms in the two chains run in opposite directions. Each chain loosely resembles Furberg’s (2) model No. 1; that is, the bases are on the inside of the helix and the phosphates on the outside. The configuration of the sugar and the atoms near it is close to Furberg’s ‘standard configuration’, the sugar being roughly perpendicular to the attached base. There is a residue on each chain every 3.4 A. in the z-direction. We have assumed an angle of 36° between adjacent residues in the same chain, so that the structure repeats after 10 residues on each chain, that is, after 34 A. The distance of a phosphorus atom from the fibre axis is 10 A. As the phosphates are on the outside, cations have easy access to them.
View larger version (16K):
[in this window]
[in a new window]
This figure is purely diagrammatic. The two ribbons symbolize the two phosphate-sugar chains, and the horizontal rods the pairs of bases holding the chains together. The vertical line marks the fibre axis.
The structure is an open one, and its water content is rather high. At lower water contents we would expect the bases to tilt so that the structure could become more compact.
The novel feature of the structure is the manner in which the two chains are held together by the purine and pyrimidine bases. The planes of the bases are perpendicular to the fibre axis. They are joined together in pairs, a single base from one chain being hydrogen-bonded to a single base from the other chain, so that the two lie side by side with identical z-co-ordinates. One of the pair must be a purine and the other a pyrimidine for bonding to occur. The hydrogen bonds are made as follows: purine position 1 to pyrimidine position 1; purine position 6 to pyrimidine position 6.
If it is assumed that the bases only occur in the structure in the most plausible tautomeric forms (that is, with the keto rather than the enol configurations) it is found that only specific pairs of bases can bond together. These pairs are: adenine (purine) with thymine (pyrimidine), and guanine (purine) with cytosine (pyrimidine).
In other words, if an adenine forms one member of a pair, on either chain, then on these assumptions the other member must be thymine; similarly for guanine and cytosine. The sequence of bases on a single chain does not appear to be restricted in any way. However, if only specific pairs of bases can be formed, it follows that if the sequence of bases on one chain is given, then the sequence on the other chain is automatically determined.
It has been found experimentally (3, 4) that the ratio of the amounts of adenine to thymine, and the ratio of guanine to cytosine, are always very close to unity for deoxyribose nucleic acid.
It is probably impossible to build this structure with a ribose sugar in place of the deoxyribose, as the extra oxygen atom would make too close a van der Waals contact. The previously published X-ray data (5, 6) on deoxyribose nucleic acid are insufficient for a rigorous test of our structure. So far as we can tell, it is roughly compatible with the experimental data, but it must be regarded as unproved until it has been checked against more exact results. Some of these are given in the following communications. We were not aware of the details of the results presented there when we devised our structure, which rests mainly though not entirely on published experimental data and stereochemical arguments.
It has not escaped our notice that the specific pairing we have postulated immediately suggests a possible copying mechanism for the genetic material.
Full details of the structure, including the conditions assumed in building it, together with a set of co-ordinates for the atoms, will be published elsewhere.
Footnotes
We are much indebted to Dr. Jerry Donohue for constant advice and criticism, especially on interatomic distances. We have also been stimulated by a knowledge of the general nature of the unpublished experimental results and ideas of Dr. M.H.F. Wilkins, Dr. R.E. Franklin and their co-workers at King"s College, London. One of us (J.D.W.) has been aided by a fellowship from the National Foundation for Infantile Paralysis.
References
1. Pauling, L., and Corey, R. B., Nature, 171, 346 (1953); Proc. U.S. Nat. Acad. Sci., 39, 84 (1953).
2. Furberg, S., Acta Chem. Scand., 6, 634 (1952).
3. Chargaff, E., for references see Zamenhof, S., Brawerman, G., and Chargaff, E., Biochim. et Biophys. Acta, 9, 402 (1952).
4. Wyatt, G. R., J. Gen. Physiol., 36, 201 (1952).
5. Astbury, W. T., Symp. Soc. Exp. Biol. 1, Nucleic Acid, 66 (Camb. Univ. Press, 1947).
6. Wilkins, M. H. F., and Randall, J. T., Biochim. et Biophys. Acta, 10, 192 (1953).
Subscribe to:
Posts (Atom)